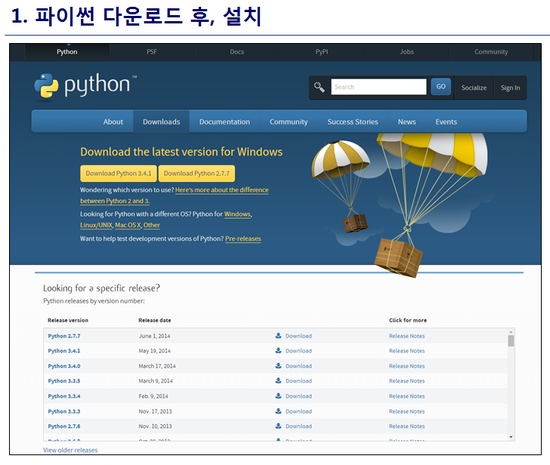
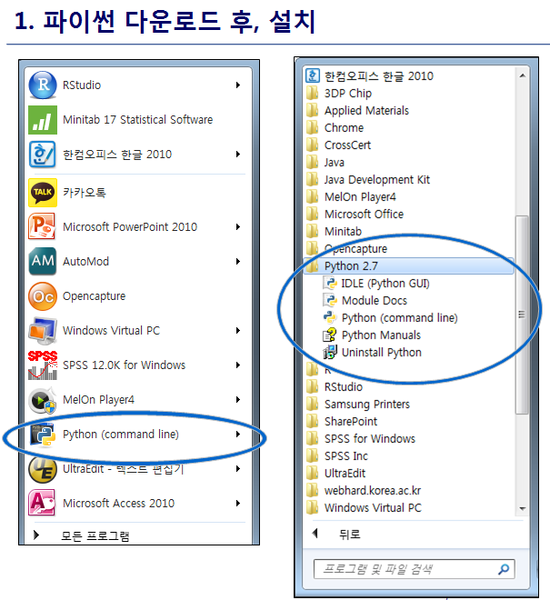
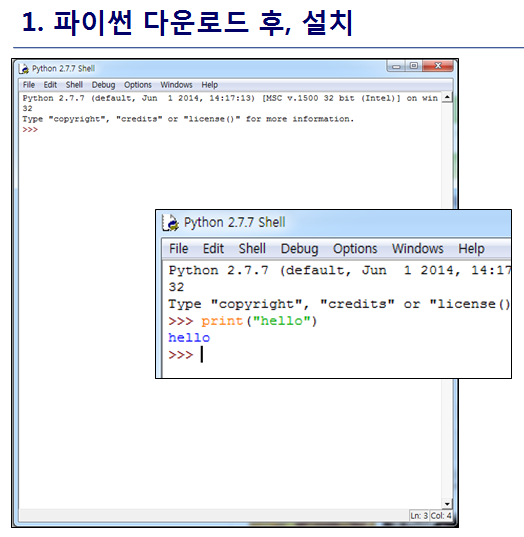
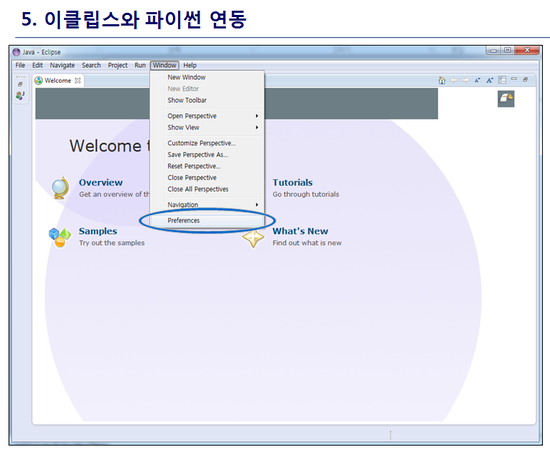
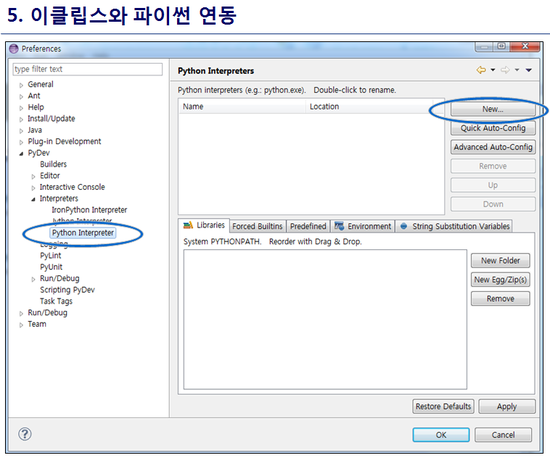
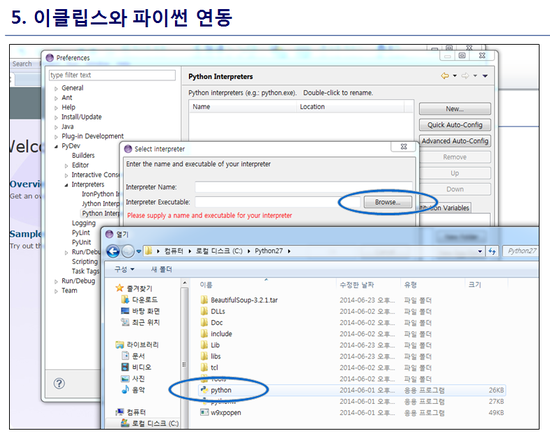
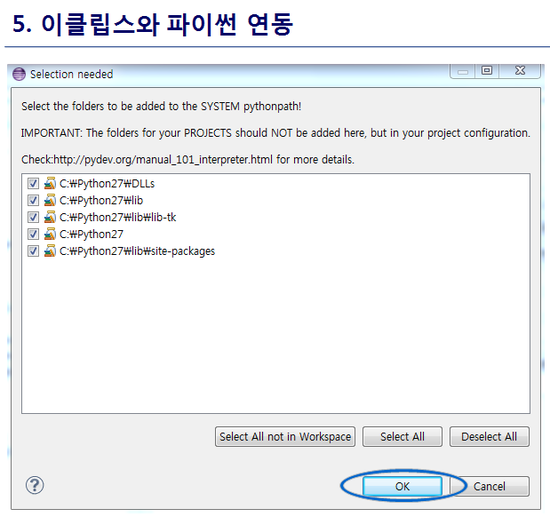
최신 버전 활용
1. Python 3.8 설치
출처: https://codechacha.com/ko/install-python37-in-ubuntu1804/
> wget www.python.org/ftp/python/3.8.2/Python-3.8.2.tgz
> tar -zxvf Python-3.8.2.tgz
> cd Python-3.8.2
> ./configure
> make
> sudo make install
Python 2/3 을 혼용으로 쓰고 싶을 때 Alternative 활용!
출처: https://codechacha.com/ko/change-python-version/
> sudo update-alternatives --config python
결과: update-alternatives: error: no alternatives for python
> sudo update-alternatives --install /usr/bin/python python /usr/bin/python2.7 1
> sudo update-alternatives --install /usr/bin/python python /usr/local/bin/python3.8 2
> sudo update-alternatives --config python
결과: There are 2 choices for the alternative python (providing /usr/bin/python).
Selection Path Priority Status
------------------------------------------------------------
* 0 /usr/bin/python3.6 2 auto mode
1 /usr/bin/python2.7 1 manual mode
2 /usr/bin/python3.6 2 manual mode
2. boost 1.73 설치 with Python3
출처: https://gist.github.com/melvincabatuan/a5a4a10b15ef31a5a481
1. Check Python3 root
>>> import sys
>>> import os
>>> sys.executable '/usr/local/bin/python3'
OR $ which python3 /usr/local/bin/python3
2. Modify user-config.jam
Ex. location:
/boost_1_57_0/tools/build/example/user-config.jam
Ex. modification
using python : 3.4 : /usr/local/bin/python3 : /usr/local/include/python3.4m : /usr/local/lib ;
4. sudo ./bootstrap.sh --with-python=/usr/local/bin/python3 --with-python-version=3.8 --with-python-root=/usr/local/include/python3.8
Ex.
Building Boost.Build engine with toolset gcc... tools/build/src/engine/bin.linuxx86/b2 Unicode/ICU support for Boost.Regex?... /usr Backing up existing Boost.Build configuration in project-config.jam.3 Generating Boost.Build configuration in project-config.jam... Bootstrapping is done. To build, run: ./b2 To adjust configuration, edit 'project-config.jam'. Further information: - Command line help: ./b2 --help - Getting started guide: http://www.boost.org/more/getting_started/unix-variants.html - Boost.Build documentation: http://www.boost.org/boost-build2/doc/html/index.html
5. sudo ./b2 --enable-unicode=ucs4 install (<- 반드시 이렇게 설치할 것)
$ sudo ./b2 --enable-unicode=ucs4 install
sudo: unable to resolve host cobalt [sudo] password for cobalt: Performing configuration checks - 32-bit : yes (cached) - arm : no (cached) - mips1 : no (cached) - power : no (cached) - sparc : no (cached) - x86 : yes (cached) - lockfree boost::atomic_flag : yes (cached) - has_icu builds : yes (cached) warning: Graph library does not contain MPI-based parallel components. note: to enable them, add "using mpi ;" to your user-config.jam - zlib : yes (cached) - iconv (libc) : yes (cached) - icu : yes (cached) - compiler-supports-ssse3 : yes (cached) - compiler-supports-avx2 : yes (cached) - gcc visibility : yes (cached) - long double support : yes (cached) warning: skipping optional Message Passing Interface (MPI) library. note: to enable MPI support, add "using mpi ;" to user-config.jam. note: to suppress this message, pass "--without-mpi" to bjam. note: otherwise, you can safely ignore this message. - zlib : yes (cached) Component configuration: - atomic : building - chrono : building - container : building - context : building - coroutine : building - date_time : building - exception : building - filesystem : building - graph : building - graph_parallel : building - iostreams : building - locale : building - log : building - math : building - mpi : building - program_options : building - python : building - random : building - regex : building - serialization : building - signals : building - system : building - test : building - thread : building - timer : building - wave : building ...patience... ...patience... ...patience... ...patience... ...patience... ...patience... ...found 35166 targets... ...updating 6 targets... gcc.link.dll bin.v2/libs/python/build/gcc-4.8/release/threading-multi/libboost_python3.so.1.57.0 common.copy /usr/local/lib/libboost_python3.so.1.57.0 ln-UNIX /usr/local/lib/libboost_python3.so gcc.archive bin.v2/libs/python/build/gcc-4.8/release/link-static/threading-multi/libboost_python3.a common.copy /usr/local/lib/libboost_python3.a ...updated 6 targets...
3. cmake 3.17.2 설치
출처: https://tttsss77.tistory.com/77
> wget https://cmake.org/files/v3.17/cmake-3.17.2.tar.gz
> tar -zxvf cmake-3.17.2.tar.gz
> cd cmake-3.17.2/
> ./bootstrap
> make
> sudo make install
install and use eigen3 on ubuntu 16.04
출처: https://kezunlin.me/post/d97b21ee/
> sudo apt-get install libeigen3-dev
4. RDKit 2020.03.2 설치
출처: https://www.rdkit.org/docs/Install.html#
Download from github.com/rdkit/rdkit/archive/Release_2020_03_2.tar.gz
> tar -zxvf Release_2020_03_2.tar.gz
> cd rdkit-Release_2020_03_2
> vi ~/.bahrc
추가:
export RDBASE=/mnt/hdd0/SKBP/lib/RDKit/rdkit-Release_2020_03_2
export LD_LIBRARY_PATH=${LD_LIBRARY_PATH}:${RDBASE}/lib:/usr/lib/x86_64-linux-gnu
export PYTHONPATH=${PYTHONPATH}:${RDBASE}
> cd $RDBASE
> mkdir build
> cd build
> cmake -D Boost_NO_BOOST_CMAKE=ON -D BOOST_ROOT=/mnt/hdd0/SKBP/lib/Boost/boost_1_73_0 -D PYTHON_LIBRARY=/usr/local/lib/python3.8/config-3.8-x86_64-linux-gnu/libpython3.8.a -D PYTHON_INCLUDE_DIR=/usr/local/include/python3.8/ -D PYTHON_EXECUTABLE=/usr/local/bin/python3 ..
> make
> sudo make install